ENFERMEDADES PRODUCIDAS POR PRIONES. (I)
Torres, JM; Brun, A; Castilla, J. y Sánchez-Vizcaíno, JM.
Propiedades físico-químicas de los priones.
En 1982 Stanley B. Prusiner propuso el nombre de
"prión" para el agente causante de un grupo de enfermedades degenerativas del
sistema nervioso central caracterizadas por ser patologías crónicas y progresivas. En
1997, Stanley B. Prusiner fue galardonado con el premio Nobel de fisiología y medicina por los
trabajos llevados a cabo para la identificación de agente infeccioso de las encefalopatías
espongiformes transmisibles (EETs). Estas enfermedades incluyen la encefalopatía espongiforme
bovina (BSE), el scrapie de las ovejas y cabras así como la enfermedad de Creutzfeldt-Jakob y su
nueva variante en seres humanos.
Los priones son pequeñas partículas infecciosas de naturaleza proteica con unas sorprendentes propiedades que las hacen más resistentes que la mayoría de las proteínas a la inactivación por métodos físico-químicos. La naturaleza proteica de los priones se sugirió dada su resistencia a la inactivación por procedimientos que destruyen los ácidos nucleicos, como por ejemplo la radiación ultravioleta. Hasta la fecha la asociación de algún tipo de ácido nucleico específico con los priones no se ha confirmado formalmente. A pesar de todo la controversia científica existe entre aquellos defensores de la teoría de la proteína única y los defensores de la hipóteis del virino o de la nucleoproteína, que sostiene que el agente infeccioso consiste en un genoma formado por ácido nucleico y la proteína del prión (PrP) derivada del huésped.
De acuerdo con la hipótesis de la proteína única, los priones se componen de una proteína denominada PrPSc (isoforma scrapie de la proteína del prión). La PrPSc se produce por el plegamiento erróneo de una proteína celular de idéntica secuencia de aminoácidos, presente en prácticamente todos los tejidos del organismo, denominada PrPC (isoforma celular de la proteína del prión)

Cambio de plegamiento de PrPC a PrPSc
Este plegamiento erróneo confiere a la PrPSc dos propiedades que permiten diferenciarla de la PrPC, la resistencia parcial a la digestión por proteasas y la insolubilidad. Las causas que provocan el error en el plegamiento de la PrPC definen las formas descritas de las enfermedades producidas por priones:
1. FORMAS ESPORÁDICAS DE LA ENFERMEDAD. Son las que aparecen sin causa aparente y para las que no hay explicación en la actualidad.
2. FORMAS INFECCIOSAS. Se explican por la interacción de la PrPSc sobre la PrPC lo que provoca su transformación a PrPSc.
3. FORMAS HEREDITARIAS. Provocadas por alteraciones genéticas heredables que facilitan el plegamiento erróneo de la PrPC
Ver tabla "Historia de las enfermedades producidas por priones"
![]()
¿Cómo son los priones?
Los priones son patógenos infecciosos que difieren de las bacterias, hongos, parásitos, virus y viroides tanto en su estructura y características físico-químicas como en la enfermedad que causan. Los priones presentan importantes características que los diferencian de los virus.
|
Diferencias fundamentales entre los priones y
los virus
|
|
|
PRIONES
|
VIRUS
|
|
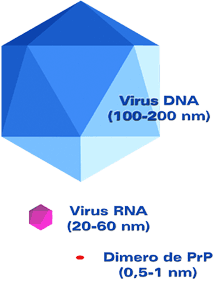
Formas moleculares múltiples (Isoformas). |
Formas únicas con diferentes morfologías estructurales |
|
No presentan ácido nucleico esencial dentro de la partícula infecciosa |
Poseen un genoma formado por ácido nucleico que sirve de molde para la replicación. |
|
No inducen respuesta inmune. |
Inducen respuesta inmune más o menos intensa según el tipo |
|
El único componente conocido es la proteína PrPSc |
Están compuestos por ácidos nucleicos, proteínas y, a menudo, por otros constituyentes. |
![]()
Propiedades físico-químicas de los priones
La masa relativa (Mr) de la PrPSc es de 33-35 x 103. Los priones se agregan en partículas sin tamaño uniforme y no pueden solubilizarse con detergentes si no es en condiciones que alteran la estructura de la PrPSc, con la consiguiente pérdida de infectividad. Sin embargo, utilizando fosfolípidos fue posible conseguir la solubilización de la PrPSc.
Los priones resisten la inactivación con nucleasas, radiación UV a 254 nm, tratamiento con psoralenos, cationes divalentes, iones metálicos quelantes, ácido (entre pH 3 y 7), hidroxilamina, formalina, ebullición y proteasas. La infectividad de los priones se disminuye mediante la digestión prolongada con proteasas, o con tratamientos como urea, ebullición en SDS, álcali (>pH 10), autoclave a 132ºC durante más de dos horas, solventes orgánicos desnaturalizantes (e. g., fenol) o agentes caotrópicos como el isocianato de guanidina.
![]()
Morfología del prión.
A partir de tejido cerebral procedente de un individuo infectado se pueden extraer vesículas membranosas ricas en PrPSc. La proteolisis parcial de estas vesículas da lugar a partículas con forma de varilla, visualizables al microscopio electrónico.
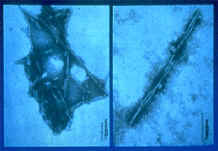
La imagen procede de http://ss.niah.affrc.go.jp/bse/images/img0035.gif
{kind=link}
La mayoría de estas partículas tienen un diámetro uniforme de 11 nm con una longitud media de 165 nm. Estas varillas son lisas, parecidas a listones, y raramente aparecen retorcidas. Las varillas están formadas por polímeros de PrPSc 27-30 (generada por la proteolisis parcial de la PrPSc 33-35).
![]()
Biología Molecular de los priones.
La proteína PrPC está codificada en un gen cromosómico muy similar entre las distintas especies de mamíferos. El gen de la PrP existe en una única copia y está localizado en el brazo corto del cromosoma 20 en el hombre y en una región equivalente del cromosoma 2 en ratones. El gen del ratón contiene tres exones, mientras que el gen humano y de hámster únicamente dos. La secuencia de aminoácidos (ORF) de todas las proteínas conocidas de priones se encuentra codificada en su totalidad dentro de un único exón.
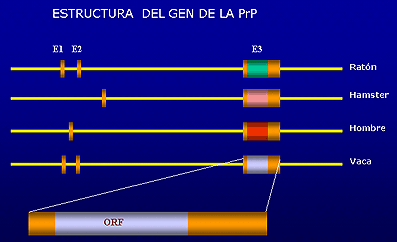
El gen se expresa de manera constitutiva prácticamente en todos los tejidos del organismo adulto siendo su expresión máxima en tejidos neuronales, fundamentalmente en el cerebro, cerebelo, médula e hipotálamo.
La PrPC tiene un tamaño comprendido entre los 33 y 35 kDa (PrP 33-35). El suceso crítico en la patogénesis parece estar relacionado con un cambio estructural que transforma a la PrPC en la PrPSc, el componente principal de las partículas infecciosas. Por tanto, ambas formas proteicas poseen idéntica secuencia de aminoacidos y la única diferencia entre ellas estriba en su estructura secundaria, es decir en la forma de plegamiento de la cadena polipeptídica. Estas diferencias conformacionales entre ambas isoformas confiere a la PrPSc las propiedades que permiten diferenciarla de la PrPC: mientras que la PrPC es digerida completamente por proteasa, la PrPSc es parcialmente resistente a esta digestión, resultado una proteína de 27 a 30 kDa (PrP 27-30).
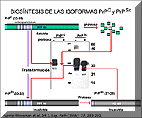
Biosíntesis de las isoformas PrPC y PrPSc
El prión es pues una forma alterada de una proteína celular normal que ha podido perder su función pero que ha adquirido la capacidad de transformar la forma normal en patológica. La patología se manifiesta probablemente por la acumulación de la proteína anormal.
![]()
Estructura de la proteína del prión.
La proteína celular madura, purificada en su forma nativa, tiene una estructura secundaria compuesta por un 43% de α-hélice y un 3% de lámina β. Por el contrario, los estudios espectroscópicos permitieron establecer que la estructura secundaria de la PrPSc contiene un 43 % de lámina β lo que le confiere la capacidad de formar complejos supramoleculares acumulables denominados amiloides.
La estructura terciaria de la proteína recombinante del prión humano se ha desvelado recientemente. Ésta consiste en tres α-hélices que conforman un núcleo ordenado en el extremo carboxilo terminal de la molécula y una zona amino terminal desestructurada y flexible.

Se ha propuesto que la transformación de PrPSc a PrPSc consiste en una transición de las α-hélices a láminas β, coincidiendo con las zonas conservadas de la cadena polipeptídica.
Sin embargo, se desconocen muchos detalles sobre el mecanismo de acción del proceso de transformación. Se ha sugerido que las dos isoformas deben interaccionar entre ellas debido a que:
La transmisión de los priones precisa de cierta especificidad entre las secuencias interaccionantes
Fue posible generar moléculas parecidas a la PrPSc in vitro, mezclando PrPC purificada con PrPSc o con péptidos sintéticos.
![]()
¿Cómo afectan los priones a los animales y al hombre?
Los priones causan enfermedades de tipo neurodegenerativo denominadas clínicamente como encefalopatías espongiformes transmisibles (EETs) (Tabla 2). Los signos clínicos y la patología que producen varían dependiendo de la especie afectada e incluso de cada animal afectado. En todos los casos el desarrollo de la enfermedad es muy lento y los tiempos de incubación, con ausencia total de síntomas, son extremadamente largos (2-10 años dependiendo de la especie y de los individuos). En general, en los animales se pueden distinguir dos fases: (Tabla 3)
FASE PSÍQUICA: se producen cambios en el comportamiento y el temperamento
En el hombre los primeros síntomas son de origen psíquico afectando a la personalidad y al comportamiento, con aparición de trastornos de memoria. A medida que la enfermedad avanza aparecen dolores musculares en las extremidades inferiores. En una fase final los síntomas principales son demencia y diestesia. La muerte sobreviene tras 6-12 meses desde la aparición de los primeros síntomas. En la actualidad es una enfermedad incurable.
Desde el punto de vista anatomopatológico, se puede decir que, macroscópicamente, los cerebros de los animales infectados aparecen normales, mientras que microscópicamente los cambios más notorios consisten en astrogliosis, vacuolización intracelular, pérdida de neuronas y formación de placas amiloides ocasionales.
![]()
¿Qué son los priones?- ¿Cómo son los priones?- Propiedades físico-químicas de los priones.- Morfología del prión.- Biología Molecular de los priones.- Estructura de la proteína del prión. -¿Cómo afectan los priones a los animales y al hombre?- Encefalopatía espongiforme de ovejas y cabras (scrapie).- Encefalopatía espongiforme bovina (BSE).-Encefalopatía transmisible del visón (TME).-Enfermedad del desgaste crónico (CWD).-Encefalopatía espongiforme felina (FSE).-¿Cómo se transmiten los priones?-Modelos propuestos para la propagación de los priones.-¿En qué consiste la barrera interespecífica de los priones?-Epidemiología y transmisión de algunas ETT´s.-¿Cómo se diagnostican las enfermedades producidas por priones?-¿Cuál es el riesgo actual de infección en el hombre?
Accede. Internet al servicio de la Ciencia y la Tecnología, ha diseñado este artículo web y sus animaciones.